

Há 50 anos, pensava-se que a quantidade de DNA em um genoma tinha uma correlação positiva com a complexidade de um organismo, ou seja, quanto mais complexa fosse uma espécie, mais DNA era necessário para armazenar aquelas informações que seriam traduzidas em fenótipos hierarquicamente mais complexos. O que pensamos hoje dessa ideia?
O conhecimento acumulado, particularmente sobre genomas e diversidade, nos permite rejeitar essa hipótese de correlação entre quantidade de DNA e complexidade. Um dos principais problemas com essa hipótese é o uso de um conceito ambíguo como complexidade. Talvez, a melhor definição – ou pelo menos mais clara – tenha sido dada há 10 anos, como sendo o número de células diferentes e o nível de organização celular de um organismo. Mas usando esta, ou qualquer uma das definições de complexidade, a alegação de correlação não é suportada pelas observações de inúmeros genomas (Figura 1). Por exemplo, se compararmos a distribuição da quantidade de DNA em eucariotos, vemos que há uma variação de várias ordens de magnitude: vai de 0,0023 picogramas no fungo parasita Encephalitozoon intestinalis até 1400 picogramas na ameba Chaos chaos, mais de 600 mil vezes maior. Para se ter uma ideia, uma célula humana tem 3,5 picogramas de DNA (1 picograma = 978 Mega bases).

Ao investigar a complexidade dos genomas em diferentes espécies, cientistas descobriram que grande parte desse DNA não era traduzido, ou seja, não carregava informação para sintetizar uma proteína. Logo descobriram que genomas muito grandes possuíam uma enorme quantidade de sequências repetitivas, padrões reconhecíveis na sequência de DNA que ocorrem em várias cópias no genoma, dispersas ou em tandem. Essas sequências estão presentes em todos os genomas e podem ter diversas origens. O genoma humano, por exemplo, incorporou ao longo do tempo DNA viral de infecções antigas. A fração do genoma correspondente a esses resquícios virais é maior que a fração do genoma que é traduzida em proteínas. Como muitas dessas sequências não possuíam nenhuma função aparente, acabaram recebendo o nome de DNA lixo quando foram descobertas. Aos poucos, foram sendo atribuídas funções a uma parte pequena dessas sequências. Elas podem ter função estrutural na organização dos cromossomos na célula e uma função regulatória no ajuste da quantidade de RNA mensageiro produzido por genes próximos (regulação da expressão gênica) [um post anterior faz uma discussão sobre outros possíveis papeis das regiões repetitivas].
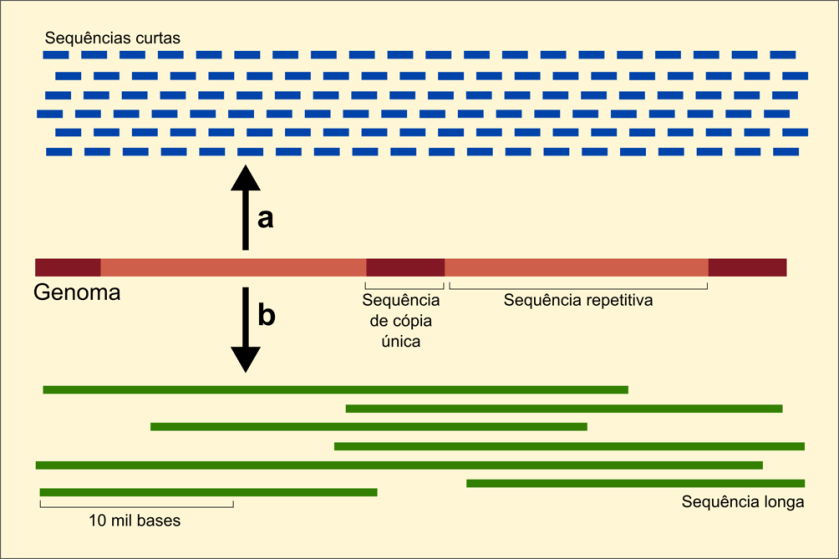
Mesmo com a atenção recebida recentemente, o avanço na compreensão do papel dessas regiões é muito mais lento que para o resto do genoma, pois elas são muito mais difíceis de se obter durante o sequenciamento de um genoma. Os métodos de sequenciamento de DNA mais comuns geram sequências que têm entre 100 e 150 nucleotídeos de comprimento. Por essa razão, o primeiro passo para sequenciar um genoma é quebrá-lo em pedaços menores para serem sequenciados. São então analisados em programas que buscam regiões de sobreposição entre eles para combiná-los e formar sequências contíguas maiores (figura 2). Em genomas ricos em regiões repetidas, muitas sobreposições são encontradas para repetições, mesmo aquelas que estão localizadas a grandes distâncias no genoma. Isso faz com que essas regiões não sejam propriamente montadas por esses programas. Podemos pensar nesse método como a montagem de um grande quebra-cabeças, com bilhões de pequenas peças, sendo muitas delas exatamente iguais.
Dois trabalhos recentes abriram a porta para novos estudos sobre a natureza, função e evolução de regiões repetitivas: o sequenciamento dos genomas do axolote, Ambystoma mexicanum, uma salamandra mexicana, e da planária Schmidtea mediterranea. O genoma do axolote com 32 bilhões de pares de bases, 10 vezes maior que o genoma humano, é o maior genoma sequenciado até hoje. Dentre as salamandras nem é tão impressionante assim; algumas espécies chegam a ter genomas 40 vezes maiores que o nosso. O genoma de S. mediterranea, por sua vez, tem cerca de 800 milhões de bases distribuídas em quatro cromossomos, menos de um terço do genoma humano. Mesmo assim, as versões anteriores de montagens do genoma dessa espécie eram muito fragmentadas em decorrência da alta frequência de sequências repetitivas, principalmente de retrotransposons, que se integraram e se replicaram em seu genoma ao longo de sua história evolutiva. Os retrotransposons usam um sistema de copiar e colar, no qual geram cópias próprias de RNA e usam uma enzima, chamada transcriptase reversa para converter seu RNA em DNA e reintegrar essa nova cópia no genoma.
Esses dois genomas foram sequenciados utilizando uma estratégia relativamente nova, que gera longas sequências de DNA, em geral maiores que 1500 bases. No entanto, a qualidade dessas sequências é baixa e sujeita a inferência incorreta de bases. Esse problema foi resolvido com a combinação de dois métodos de sequenciamento: o sequenciamento de cópia única, que gera essas sequências maiores, mas de qualidade baixa, e um segundo método que gera sequências muito curtas de boa qualidade, para corrigir as sequências maiores.
O genoma de ambos os organismos foi sequenciado para investigar possíveis bases moleculares da regeneração. O axolote tem a capacidade de regenerar órgão internos e membros amputados (incluindo ossos, músculos e nervos). Até a medula espinhal pode ser recuperada de danos e ter sua função restaurada como se nada tivesse acontecido. As planárias também são tidas como modelos biológicos para estudos de regeneração. Nelas, células tronco adultasÓtimo, pluripotentes, os neoblastos, garantem a regeneração de qualquer tipo celular. Será que esses genomas guardam segredos sobre a incrível capacidade de regeneração desses animais?
Na planária, os cientistas identificaram cerca de mil genes espécie-específicos, ou seja, esses genes não foram observados em nenhum outro genoma sequenciado. Por outro lado, centenas de genes essenciais para camundongos e humanos não aparecem no genoma da planária. Dentre eles um gene de controle do ciclo celular, e um gene essencial no metabolismo de gordura. No axolote, foram encontrados cinco genes novos, que não haviam sido observados em genomas de répteis ou mamíferos e estão ativos em membros em processo de regeneração. Destes, dois já eram conhecidos e os outros ainda precisam ser investigados. Outra observação interessante foi a evidência de que Pax3, um gene essencial para o desenvolvimento embrionário de muitos vertebrados, tenha sido perdido no axolote. É possível que Pax7, um gene relacionado, compense a ausência de Pax3, assumindo seu papel durante o desenvolvimento.
Essas observações de ganho e perda de genes, no entanto, não explicam o tamanho desses genomas. No axolote, as sequências repetitivas perfazem 65,6% do genoma (18,6 Gb). Grande parte dessas sequências consiste de cópias de retroelementos e retrovirus. Na planária foi encontrado o maior elemento de DNA móvel encontrado fora do reino Plantae, chamado Ogre. A versão da planária foi chamada de Burro, acrônimo do inglês “big, unknown repeat rivaling Ogre” (grande elemento desconhecido rival do Ogre).
Mesmo não fornecendo muitas respostas sobre as bases moleculares da regeneração, esses novos esforços de sequenciamento mostraram que é possível gerar informações para genomas ricos em repetições. Foi um passo importante para permitir a investigação da função e evolução da porção não codificante do genoma de inúmeras espécies. Por exemplo, considere o DNA repetitivo que impedia a montagem dos genomas. Em ambos os casos, no axolote e na planária, grande parte das repetições consiste de cópias de retrotransposons. Estudos anteriores sugeriram que retrotransposons poderiam participar de processos biológicos que moldam o desenvolvimento embrionário [9]. Em uma outra salamandra, Pleurodeles waltl, um transposon abundante em seu genoma foi encontrado ativo em membros em regeneração e pode ter um papel da regulação da expressão de genes envolvidos no processo. Essa é uma das hipóteses que poderão ser testadas com os novos genomas, em combinação com as técnicas de edição, contribuindo para dar suporte ou falsear a ideia de que, para genomas, tamanho não é documento.
Tatiana Teixeira Torres (USP)
Para saber mais:
Carareto CMA, Monteiro-Vitorello CB, Van Sluys MA (2015) Elementos de Transposição: Diversidade, evolução, aplicações e impacto nos genomas dos seres vivos. Rio de Janeiro: Sociedade Brasileira de Genética, FIOCRUZ; 196p.
Biscotti MA, Olmo E, Heslop-Harrison JS (2015). Repetitive DNA in eukaryotic genomes. Chromosome Res, 23(3):415-20.

Notícias sobre avanços na genética estão cada vez mais em evidência, porém, antes de compreender as novas técnicas e aplicações do universo da genética é importante compreender a base de tudo: o DNA. Nos tópicos a seguir vamos explicar o que é DNA, do que ele é composto e como o DNA é encontrado nas células do corpo humano.
CurtirCurtir